
AMP-activated protein kinase
| AMP-activated protein kinase (AMPK) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
 AMP-activated protein kinase | |||||||||
| Identifiers | |||||||||
| EC no. | 2.7.11.1 | ||||||||
| CAS no. | 172522-01-9 | ||||||||
| Alt. names | AMP-activated protein kinase | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / QuickGO | ||||||||
| |||||||||
5' AMP-activated protein kinase or AMPK or 5' adenosine monophosphate-activated protein kinase is an enzyme (EC 2.7.11.31) that plays a role in cellular energy homeostasis, largely to activate glucose and fatty acid uptake and oxidation when cellular energy is low. It belongs to a highly conserved eukaryotic protein family and its orthologues are SNF1 in yeast, and SnRK1 in plants. It consists of three proteins (subunits) that together make a functional enzyme, conserved from yeast to humans. It is expressed in a number of tissues, including the liver, brain, and skeletal muscle. In response to binding AMP and ADP,[1] the net effect of AMPK activation is stimulation of hepatic fatty acid oxidation, ketogenesis, stimulation of skeletal muscle fatty acid oxidation and glucose uptake, inhibition of cholesterol synthesis, lipogenesis, and triglyceride synthesis, inhibition of adipocyte lipogenesis, inhibition of adipocyte lipolysis, and modulation of insulin secretion by pancreatic β-cells.[2]
It should not be confused with cyclic AMP-activated protein kinase (protein kinase A).[3]
Structure
[edit]AMPK is a heterotrimeric protein complex that is formed by α, β, and γ subunits. Each of these three subunits takes on a specific role in both the stability and activity of AMPK.[4][5] Specifically, the γ subunit includes four particular Cystathionine-β-synthase (CBS) domains, giving AMPK its ability to sensitively detect shifts in the AMP/ATP ratio. AMPK is deactivated upon AMP displacement by ATP at CBS site 3, suggesting CBS3 to be the primary allosteric regulatory site.[6][7][8] The four CBS domains create two binding sites for AMP commonly referred to as Bateman domains. Binding of one AMP to a Bateman domain cooperatively increases the binding affinity of the second AMP to the other Bateman domain.[9][failed verification] As AMP binds both Bateman domains the γ subunit undergoes a conformational change which exposes the catalytic domain found on the α subunit. It is in this catalytic domain where AMPK becomes activated when phosphorylation takes place at threonine-172 (on α1 isoform) or Thr-174 (on α2 isoform) by an upstream AMPK kinase (AMPKK).[10][6] The α, β, and γ subunits can also be found in different isoforms: the γ subunit can exist as either the γ1, γ2 or γ3 isoform; the β subunit can exist as either the β1 or β2 isoform; and the α subunit can exist as either the α1 or α2 isoform. Although the most common isoforms expressed in most cells are the α1, β1, and γ1 isoforms, it has been demonstrated that the α2, β2, γ2, and γ3 isoforms are also expressed in cardiac and skeletal muscle.[4][11][12]
The following human genes encode AMPK subunits:
The crystal structure of mammalian AMPK regulatory core domain (α C terminal, β C terminal, γ) has been solved in complex with AMP,[13] ADP [14] or ATP.[15]
Regulation
[edit]Due to the presence of isoforms of its components, there are 12 versions of AMPK in mammals, each of which can have different tissue localizations, and different functions under different conditions.[16] AMPK is regulated allosterically and by post-translational modification, which work together.[16]
If residue Thr-172 of AMPK's α1-subunit (or Thr-174 of AMPK's α2-subunit) is phosphorylated, AMPK is activated around 100-fold;[6] access to that residue by phosphatases is blocked if AMP or ADP can block access for and ATP can displace AMP and ADP.[16] That residue is phosphorylated by at least three kinases (liver kinase B1 (LKB1),[17] which works in a complex with STRAD and MO25, Calcium/calmodulin-dependent protein kinase kinase II-(CAMKK2), and TGFβ-activated kinase 1 (TAK1)) and is dephosphorylated by three phosphatases (protein phosphatase 2A (PP2A); protein phosphatase 2C (PP2C) and Mg2+-/Mn2+-dependent protein phosphatase 1E (PPM1E)).[16]
Regulation of AMPK by CaMKK2 requires a direct interaction of these two proteins via their kinase domains. The interaction of CaMKK2 with AMPK only involves the α and β subunits of AMPK (AMPK γ is absent from the CaMKK2 complex), thus rendering regulation of AMPK in this context to changes in calcium levels but not AMP or ADP.
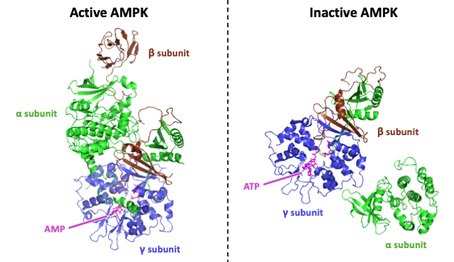
AMPK is regulated allosterically mostly by competitive binding to the CBS sites on its γ subunit between ATP (which allows phosphatase access to Thr-172) and AMP or ADP (each of which blocks access to phosphatases).[1] It thus appears that AMPK is a sensor of AMP/ATP or ADP/ATP ratios and thus cell energy level.[16] AMPK undergoes a large conformational change upon ATP binding. A region on the α subunit known as the kinase domain (KD) dissociates from its active-state conformation and loosely associates with the γ subunit ~100Å away. The KD also rotates ~180° in the conformational change. Upon KD dissociation, the active loop (AL) of the α subunit which contains the critical phosphorylated Thr residue is fully exposed to upstream phosphatases. This conformational change represents a plausible mechanism for AMPK modulation. When cellular energy states are low (high AMP/ATP or ADP/ATP levels), AMPK adopts the KD-associated conformation and AMPK is protected from dephosphorylation and remains activated. When cellular energy states are high, AMPK adopts the KD-displaced conformation, the AL is exposed to upstream phosphatases, and AMPK is deactivated.[6]
The pharmacological compounds Merck Compound 991 and Abbott A769662 bind to the allosteric drug and metabolism site (ADaM) on the β subunit and have been shown to activate AMPK up to 10-fold.[6][18] ADaM site binding may have roles in AMPK activation as well as protection against dephosphorylation.[19]
There are other mechanisms by which AMPK is inhibited or activated by insulin, leptin, and diacylglycerol by inducing various other phosphorylations.[16][a]
AMPK may be inhibited or activated by various tissue-specific ubiquitinations.[16]
It is also regulated by several protein-protein interactions, and may either be activated or inhibited by oxidative factors; the role of oxidation in regulating AMPK was controversial as of 2016.[16]
Function
[edit]When AMPK phosphorylates acetyl-CoA carboxylase 1 (ACC1) or sterol regulatory element-binding protein 1c (SREBP1c), it inhibits synthesis of fatty acids, cholesterol, and triglycerides, and activates fatty acid uptake and β-oxidation.[16]
AMPK stimulates glucose uptake in skeletal muscle by phosphorylating Rab-GTPase-activating protein TBC1D1, which ultimately induces fusion of GLUT4 vesicles with the plasma membrane.[16] AMPK stimulates glycolysis by activating phosphorylation of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2/3 and activating phosphorylation of glycogen phosphorylase, and it inhibits glycogen synthesis through inhibitory phosphorylation of glycogen synthase.[16] In the liver, AMPK inhibits gluconeogenesis by inhibiting transcription factors including hepatocyte nuclear factor 4 (HNF4) and CREB regulated transcription coactivator 2 (CRTC2).[16]
AMPK inhibits the energy-intensive protein biosynthesis process and can also force a switch from cap-dependent translation to cap-independent translation, which requires less energy, by phosphorylation of TSC2, RPTOR, transcription initiation factor 1A.66, and eEF2K.[16] When TSC2 is activated it inhibits mTORC1. As a result of inhibition of mTORC1 by AMPK, protein synthesis comes to a halt. Activation of AMPK signifies low energy within the cell, so all of the energy consuming pathways like protein synthesis are inhibited, and pathways that generate energy are activated to restore appropriate energy levels in the cell.[20]
AMPK activates autophagy by directly and indirectly activating ULK1.[16] AMPK also appears to stimulate mitochondrial biogenesis by regulating PGC-1α which in turn promotes gene transcription in mitochondria.[16] AMPK also activates anti-oxidant defenses.[16]
Clinical significance
[edit]Exercise/training
[edit]Many biochemical adaptations of skeletal muscle that take place during a single bout of exercise or an extended duration of training, such as increased mitochondrial biogenesis and capacity,[21][22] increased muscle glycogen,[23] and an increase in enzymes which specialize in glucose uptake in cells such as GLUT4 and hexokinase II [24][25] are thought to be mediated in part by AMPK when it is activated.[26][27] Additionally, recent discoveries can conceivably suggest a direct AMPK role in increasing blood supply to exercised/trained muscle cells by stimulating and stabilizing both vasculogenesis and angiogenesis.[28] Taken together, these adaptations most likely transpire as a result of both temporary and maintained increases in AMPK activity brought about by increases in the AMP:ATP ratio during single bouts of exercise and long-term training.
During a single acute exercise bout, AMPK allows the contracting muscle cells to adapt to the energy challenges by increasing expression of hexokinase II,[23] translocation of GLUT4 to the plasma membrane,[29][30][31][32] for glucose uptake, and by stimulating glycolysis.[33] If bouts of exercise continue through a long-term training regimen, AMPK and other signals will facilitate contracting muscle adaptations by escorting muscle cell activity to a metabolic transition resulting in a fatty-acid oxidation approach to ATP generation as opposed to a glycolytic approach. AMPK accomplishes this transition to the oxidative mode of metabolism by upregulating and activating oxidative enzymes such as hexokinase II, PPAR-α, PPAR-δ, PGC-1, UCP-3, cytochrome C and TFAM.[26][23][25][34][35][36][37]
Mutations in the skeletal muscle calcium release channel (RYR1) underlies a life- threatening response to heat in patients with malignant hyperthermia susceptibility (MHS). Upon acute exposure to heat, these mutations cause uncontrolled Ca2+ release from the sarcoplasmic reticulum, leading to sustained muscle contractures, severe hyperthermia, and sudden death.[38] At basal conditions, the temperature-dependent Ca2+ leak also leads to increased energy demand and activation of energy sensing AMP kinase (AMPK) in skeletal muscle.[38] The activated AMPK increases muscle metabolic activity, including glycolysis, which leads to marked elevation of circulating lactate.[38]
AMPK activity increases with exercise and the LKB1/MO25/STRAD complex is considered to be the major upstream AMPKK of the 5’-AMP-activated protein kinase phosphorylating the α subunit of AMPK at Thr-172.[10][39][40][17] This fact is puzzling considering that although AMPK protein abundance has been shown to increase in skeletal tissue with endurance training, its level of activity has been shown to decrease with endurance training in both trained and untrained tissue.[41][42][43][44] Currently, the activity of AMPK immediately following a 2 hour bout of exercise of an endurance trained rat is unclear. It is possible that a direct link exists between the observed decrease in AMPK activity in endurance trained skeletal muscle and the apparent decrease in the AMPK response to exercise with endurance training.
Although AMPKα2 activation has been thought to be important for mitochondrial adaptations to exercise training, a recent study investigating the response to exercise training in AMPKα2 knockout mice opposes this idea.[45] Their study compared the response to exercise training of several proteins and enzymes in wild type and AMPKα2 knockout mice. And even though the knockout mice had lower basal markers of mitochondrial density (COX-1, CS, and HAD), these markers increased similarly to the wild type mice after exercise training. These findings are supported by another study also showing no difference in mitochondrial adaptations to exercise training between wild type and knockout mice.[46]
Maximum life span
[edit]The C. elegans homologue of AMPK, aak-2, has been shown by Michael Ristow and colleagues to be required for extension of life span in states of glucose restriction mediating a process named mitohormesis.[47]
Lipid metabolism
[edit]One of the effects of exercise is an increase in fatty acid metabolism, which provides more energy for the cell. One of the key pathways in AMPK's regulation of fatty acid oxidation is the phosphorylation and inactivation of acetyl-CoA carboxylase.[28] Acetyl-CoA carboxylase (ACC) converts acetyl-CoA to malonyl-CoA, an inhibitor of carnitine palmitoyltransferase 1 (CPT-1). CPT-1 transports fatty acids into the mitochondria for oxidation. Inactivation of ACC, therefore, results in increased fatty acid transport and subsequent oxidation. It is also thought that the decrease in malonyl-CoA occurs as a result of malonyl-CoA decarboxylase (MCD), which may be regulated by AMPK.[21] MCD is an antagonist to ACC, decarboxylating malonyl-CoA to acetyl-CoA, resulting in decreased malonyl-CoA and increased CPT-1 and fatty acid oxidation. AMPK also plays an important role in lipid metabolism in the liver. It has long been known that hepatic ACC has been regulated in the liver by phosphorylation.[22] AMPK also phosphorylates and inactivates 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR), a key enzyme in cholesterol synthesis.[29] HMGR converts 3-hydroxy-3-methylglutaryl-CoA, which is made from acetyl-CoA, into mevalonic acid, which then travels down several more metabolic steps to become cholesterol. AMPK, therefore, helps regulate fatty acid oxidation and cholesterol synthesis.
Glucose transport
[edit]Insulin is a hormone which helps regulate glucose levels in the body. When blood glucose is high, insulin is released from the Islets of Langerhans. Insulin, among other things, will then facilitate the uptake of glucose into cells via increased expression and translocation of glucose transporter GLUT-4.[27] Under conditions of exercise, however, blood sugar levels are not necessarily high, and insulin is not necessarily activated, yet muscles are still able to bring in glucose. AMPK seems to be responsible in part for this exercise-induced glucose uptake. Goodyear et al.[24] observed that with exercise, the concentration of GLUT-4 was increased in the plasma membrane, but decreased in the microsomal membranes, suggesting that exercise facilitates the translocation of vesicular GLUT-4 to the plasma membrane. While acute exercise increases GLUT-4 translocation, endurance training will increase the total amount of GLUT-4 protein available.[25] It has been shown that both electrical contraction and AICA ribonucleotide (AICAR) treatment increase AMPK activation, glucose uptake, and GLUT-4 translocation in perfused rat hindlimb muscle, linking exercise-induced glucose uptake to AMPK.[48][23][34] Chronic AICAR injections, simulating some of the effects of endurance training, also increase the total amount of GLUT-4 protein in the muscle cell.[35]
Two proteins are essential for the regulation of GLUT-4 expression at a transcriptional level – myocyte enhancer factor 2 (MEF2) and GLUT4 enhancer factor (GEF). Mutations in the DNA binding regions for either of these proteins results in ablation of transgene GLUT-4 expression.[30][31] These results prompted a study in 2005 which showed that AMPK directly phosphorylates GEF, but it doesn't seem to directly activate MEF2.[32] AICAR treatment has been shown, however, to increase transport of both proteins into the nucleus, as well as increase the binding of both to the GLUT-4 promoter region.[32]
There is another protein involved in carbohydrate metabolism that is worthy of mention along with GLUT-4. The enzyme hexokinase phosphorylates a six-carbon sugar, most notably glucose, which is the first step in glycolysis. When glucose is transported into the cell it is phosphorylated by hexokinase. This phosphorylation keeps glucose from leaving the cell, and by changing the structure of glucose through phosphorylation, it decreases the concentration of glucose molecules, maintaining a gradient for more glucose to be transported into the cell. Hexokinase II transcription is increased in both red and white skeletal muscle upon treatment with AICAR.[36] With chronic injections of AICAR, total protein content of hexokinase II increases in rat skeletal muscle.[49]
Mitochondria
[edit]Mitochondrial enzymes, such as cytochrome c, succinate dehydrogenase, malate dehydrogenase, α-ketoglutarate dehydrogenase, and citrate synthase, increase in expression and activity in response to exercise.[43] AICAR stimulation of AMPK increases cytochrome c and δ-aminolevulinate synthase (ALAS), a rate-limiting enzyme involved in the production of heme. Malate dehydrogenase and succinate dehydrogenase also increase, as well as citrate synthase activity, in rats treated with AICAR injections.[44] Conversely, in LKB1 knockout mice, there are decreases in cytochrome c and citrate synthase activity, even if the mice are "trained" by voluntary exercise.[50]
AMPK is required for increased peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) expression in skeletal muscle in response to creatine depletion.[51] PGC-1α is a transcriptional regulator for genes involved in fatty acid oxidation, gluconeogenesis, and is considered the master regulator for mitochondrial biogenesis.[52]
To do this, it enhances the activity of transcription factors like nuclear respiratory factor 1 (NRF-1), myocyte enhancer factor 2 (MEF2), host cell factor (HCF), and others.[9][33] It also has a positive feedback loop, enhancing its own expression.[53] Both MEF2 and cAMP response element (CRE) are essential for contraction-induced PGC-1α promoter activity.[33] LKB1 knockout mice show a decrease in PGC-1α, as well as mitochondrial proteins.[50][54]
Thyroid hormone
[edit]AMPK and thyroid hormone regulate some similar processes. Knowing these similarities, Winder and Hardie et al. designed an experiment to see if AMPK was influenced by thyroid hormone.[55][56][57] They found that all of the subunits of AMPK were increased in skeletal muscle, especially in the soleus and red quadriceps, with thyroid hormone treatment. There was also an increase in phospho-ACC, a marker of AMPK activity.[55]
Glucose sensing systems
[edit]Loss of AMPK has been reported to alter the sensitivity of glucose sensing cells, through poorly defined mechanisms. Loss of the AMPKα2 subunit in pancreatic β-cells and hypothalamic neurons decreases the sensitivity of these cells to changes in extracellular glucose concentration.[58][59][60][61] Moreover, exposure of rats to recurrent bouts of insulin induced hypoglycemia/glucopenia, reduces the activation of AMPK within the hypothalamus, whilst also suppressing the counterregulatory response to hypoglycemia. [62][63] Pharmacological activation of AMPK by delivery of AMPK activating drug AICAR, directly into the hypothalamus can increase the counterregulatory response to hypoglycaemia.[64]
Lysosomal damage, inflammatory diseases, and metformin
[edit]AMPK is recruited to lysosomes and regulated at the lysosomes via several systems of clinical significance. This includes the AXIN - LKB1 complex, acting in response to glucose limitations functioning independently of AMP sensing, which detects low glucose as absence of fructose-1,6-bisphosphate via a dynamic set of interactions between lysosomally localized V-ATPase-aldolase in contact with the endoplasmic reticulum localized TRPV.[65] A second AMPK-control system[66] localized to lysosomes depends on the Galectin-9-TAK1 system and ubiquitination responses at controlled by deubiquitinating enzymes such as USP9X leading to AMPK activation in response to lysosomal damage,[66] a condition that can occur biochemically, physically via protein aggregates such as proteopathic tau in Alzheimer's disease,[67][68] crystalline silica causing silicosis,[68] cholesterol crystals causing inflammation via NLRP3 inflammasome and rupture of atherosclerotic lesions,[69] urate crystals associated with gout, or during microbial invasion such as Mycobacterium tuberculosis[68][70] or coronaviruses causing SARS.[71] Both of the above lysosomally localized systems controlling AMPK activate it in response to metformin,[66][72] a widely prescribed anti-diabetic drug.
Tumor suppression and promotion
[edit]Some evidence indicates that AMPK may have a role in tumor suppression. Studies have found that AMPK may exert most, or even all of, the tumor suppressing properties of liver kinase B1 (LKB1).[17] Additionally, studies where the AMPK activator metformin was used to treat diabetes found a correlation with a reduced risk of cancer, compared to other medications. Gene knockout and knockdown studies with mice found that mice without the gene to express AMPK had greater risks of developing lymphomas, though as the gene was knocked out globally instead of just in B cells, it was impossible to conclude that AMP knockout had cell-autonomous effects within tumor progenitor cells.[73]
In contrast, some studies have linked AMPK with a role as a tumor promoter by protecting cancer cells from stress. Thus, once cancerous cells have formed in an organism, AMPK may swap from protecting against cancer to protecting the cancer itself. Studies have found that tumor cells with AMPK knockout are more susceptible to death by glucose starvation or extracellular matrix detachment, which may indicate AMPK has a role in preventing these two outcomes.[73] A recent study on pancreatic cancer suggests that AMPKα may play a role in the metastatic cascade and the phenotype of cancer cells. Mechanistically, the authors propose that in the absence of AMPKα, pancreatic cancer cells are more vulnerable to oxidative stress, supporting a tumor-promoting function of AMPKα. [74]
Controversy over role in adaption to exercise/training
[edit]This section is written like a personal reflection, personal essay, or argumentative essay that states a Wikipedia editor's personal feelings or presents an original argument about a topic. (January 2021) |
A seemingly paradoxical role of AMPK occurs when we take a closer look at the energy-sensing enzyme in relation to exercise and long-term training. Similar to short-term acute training scale, long-term endurance training studies also reveal increases in oxidative metabolic enzymes, GLUT-4, mitochondrial size and quantity, and an increased dependency on the oxidation of fatty acids; however, Winder et al. reported in 2002 that despite observing these increased oxidative biochemical adaptations to long-term endurance training (similar to those mentioned above), the AMPK response (activation of AMPK with the onset of exercise) to acute bouts of exercise decreased in red quadriceps (RQ) with training (3 – see Fig.1). Conversely, the study did not observe the same results in white quadriceps (WQ) and soleus (SOL) muscles that they did in RQ. The trained rats used for that endurance study ran on treadmills 5 days/wk in two 1-h sessions, morning and afternoon. The rats were also running up to 31m/min (grade 15%). Finally, following training, the rats were sacrificed either at rest or following 10 minutes of exercise.
Because the AMPK response to exercise decreases with increased training duration, many questions arise that would challenge the AMPK role with respect to biochemical adaptations to exercise and endurance training. This is due in part to the marked increases in the mitochondrial biogenesis, upregulation of GLUT-4, UCP-3, Hexokinase II along with other metabolic and mitochondrial enzymes despite decreases in AMPK activity with training. Questions also arise because skeletal muscle cells which express these decreases in AMPK activity in response to endurance training also seem to be maintaining an oxidative dependent approach to metabolism, which is likewise thought to be regulated to some extent by AMPK activity.[34][35]
If the AMPK response to exercise is responsible in part for biochemical adaptations to training, how then can these adaptations to training be maintained if the AMPK response to exercise is being attenuated with training? It is hypothesized that these adaptive roles to training are maintained by AMPK activity and that the increases in AMPK activity in response to exercise in trained skeletal muscle have not yet been observed due to biochemical adaptations that the training itself stimulated in the muscle tissue to reduce the metabolic need for AMPK activation. In other words, due to previous adaptations to training, AMPK will not be activated, and further adaptation will not occur, until the intracellular ATP levels become depleted from an even higher intensity energy challenge than prior to those previous adaptations.
See also
[edit]Notes
[edit]References
[edit]- ^ a b Suter M, Riek U, Tuerk R, Schlattner U, Wallimann T, Neumann D (October 2006). "Dissecting the role of 5'-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase". The Journal of Biological Chemistry. 281 (43): 32207–16. doi:10.1074/jbc.M606357200. PMID 16943194.
- ^ Winder WW, Hardie DG (July 1999). "AMP-activated protein kinase, a metabolic master switch: possible roles in type 2 diabetes". The American Journal of Physiology. 277 (1): E1-10. doi:10.1152/ajpendo.1999.277.1.E1. PMID 10409121.
- ^ Hallows KR, Alzamora R, Li H, Gong F, Smolak C, Neumann D, Pastor-Soler NM (April 2009). "AMP-activated protein kinase inhibits alkaline pH- and PKA-induced apical vacuolar H+-ATPase accumulation in epididymal clear cells". American Journal of Physiology. Cell Physiology. 296 (4): C672-81. doi:10.1152/ajpcell.00004.2009. PMC 2670645. PMID 19211918.
- ^ a b Stapleton D, Mitchelhill KI, Gao G, Widmer J, Michell BJ, Teh T, et al. (January 1996). "Mammalian AMP-activated protein kinase subfamily". The Journal of Biological Chemistry. 271 (2): 611–4. doi:10.1074/jbc.271.2.611. PMID 8557660.
- ^ Riek U, Scholz R, Konarev P, Rufer A, Suter M, Nazabal A, et al. (June 2008). "Structural properties of AMP-activated protein kinase: dimerization, molecular shape, and changes upon ligand binding". The Journal of Biological Chemistry. 283 (26): 18331–18343. doi:10.1074/jbc.M708379200. PMID 18372250.
- ^ a b c d e Yan Y, Mukherjee S, Harikumar KG, Strutzenberg TS, Zhou XE, Suino-Powell K, et al. (July 2021). "Structure of an AMPK complex in an inactive, ATP-bound state". Science. 373 (6553): 413–419. Bibcode:2021Sci...373..413Y. doi:10.1126/science.abe7565. PMC 8428800. PMID 34437114.
- ^ Oakhill JS, Chen ZP, Scott JW, Steel R, Castelli LA, Ling N, et al. (November 2010). "β-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK)". Proceedings of the National Academy of Sciences of the United States of America. 107 (45): 19237–19241. Bibcode:2010PNAS..10719237O. doi:10.1073/pnas.1009705107. PMC 2984171. PMID 20974912.
- ^ Chen L, Wang J, Zhang YY, Yan SF, Neumann D, Schlattner U, et al. (June 2012). "AMP-activated protein kinase undergoes nucleotide-dependent conformational changes". Nature Structural & Molecular Biology. 19 (7): 716–718. doi:10.1038/nsmb.2319. PMID 22659875. S2CID 13591617.
- ^ a b Adams J, Chen ZP, Van Denderen BJ, Morton CJ, Parker MW, Witters LA, et al. (January 2004). "Intrasteric control of AMPK via the gamma1 subunit AMP allosteric regulatory site". Protein Science. 13 (1): 155–65. doi:10.1110/ps.03340004. PMC 2286513. PMID 14691231.
- ^ a b Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG (November 1996). "Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase". The Journal of Biological Chemistry. 271 (44): 27879–87. doi:10.1074/jbc.271.44.27879. PMID 8910387.
- ^ Thornton C, Snowden MA, Carling D (May 1998). "Identification of a novel AMP-activated protein kinase beta subunit isoform that is highly expressed in skeletal muscle". The Journal of Biological Chemistry. 273 (20): 12443–50. doi:10.1074/jbc.273.20.12443. PMID 9575201.
- ^ Cheung PC, Salt IP, Davies SP, Hardie DG, Carling D (March 2000). "Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding". The Biochemical Journal. 346 (3): 659–69. doi:10.1042/0264-6021:3460659. PMC 1220898. PMID 10698692.
- ^ Xiao B, Heath R, Saiu P, Leiper FC, Leone P, Jing C, et al. (September 2007). "Structural basis for AMP binding to mammalian AMP-activated protein kinase". Nature. 449 (7161): 496–500. Bibcode:2007Natur.449..496X. doi:10.1038/nature06161. PMID 17851531. S2CID 4345919.
- ^ Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, et al. (April 2011). "Structure of mammalian AMPK and its regulation by ADP". Nature. 472 (7342): 230–3. Bibcode:2011Natur.472..230X. doi:10.1038/nature09932. PMC 3078618. PMID 21399626.
- ^ Chen L, Wang J, Zhang YY, Yan SF, Neumann D, Schlattner U, et al. (June 2012). "AMP-activated protein kinase undergoes nucleotide-dependent conformational changes". Nature Structural & Molecular Biology. 19 (7): 716–8. doi:10.1038/nsmb.2319. PMID 22659875. S2CID 13591617.
- ^ a b c d e f g h i j k l m n o p q Jeon SM (July 2016). "Regulation and function of AMPK in physiology and diseases". Experimental & Molecular Medicine. 48 (7): e245. doi:10.1038/emm.2016.81. PMC 4973318. PMID 27416781.
- ^ a b c Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, et al. (November 2003). "LKB1 is the upstream kinase in the AMP-activated protein kinase cascade". Current Biology. 13 (22): 2004–8. doi:10.1016/j.cub.2003.10.031. PMID 14614828.
- ^ Calabrese MF, Rajamohan F, Harris MS, Caspers NL, Magyar R, Withka JM, et al. (August 2014). "Structural basis for AMPK activation: natural and synthetic ligands regulate kinase activity from opposite poles by different molecular mechanisms". Structure. 22 (8): 1161–1172. doi:10.1016/j.str.2014.06.009. PMID 25066137.
- ^ Xiao B, Sanders MJ, Carmena D, Bright NJ, Haire LF, Underwood E, et al. (December 2013). "Structural basis of AMPK regulation by small molecule activators". Nature Communications. 4 (1): 3017. Bibcode:2013NatCo...4.3017X. doi:10.1038/ncomms4017. PMC 3905731. PMID 24352254.
- ^ Hardie DG, Ross FA, Hawley SA (March 2012). "AMPK: a nutrient and energy sensor that maintains energy homeostasis". Nature Reviews. Molecular Cell Biology. 13 (4): 251–62. doi:10.1038/nrm3311. PMC 5726489. PMID 22436748.
- ^ a b Bergeron R, Ren JM, Cadman KS, Moore IK, Perret P, Pypaert M, et al. (December 2001). "Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis". American Journal of Physiology. Endocrinology and Metabolism. 281 (6): E1340-6. doi:10.1152/ajpendo.2001.281.6.e1340. PMID 11701451. S2CID 21577702.
- ^ a b Zong H, Ren JM, Young LH, Pypaert M, Mu J, Birnbaum MJ, Shulman GI (December 2002). "AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation". Proceedings of the National Academy of Sciences of the United States of America. 99 (25): 15983–7. Bibcode:2002PNAS...9915983Z. doi:10.1073/pnas.252625599. PMC 138551. PMID 12444247.
- ^ a b c d Holmes BF, Kurth-Kraczek EJ, Winder WW (November 1999). "Chronic activation of 5'-AMP-activated protein kinase increases GLUT-4, hexokinase, and glycogen in muscle". Journal of Applied Physiology. 87 (5): 1990–5. doi:10.1152/jappl.1999.87.5.1990. PMID 10562646. S2CID 9622267.
- ^ a b Ojuka EO, Jones TE, Nolte LA, Chen M, Wamhoff BR, Sturek M, Holloszy JO (May 2002). "Regulation of GLUT4 biogenesis in muscle: evidence for involvement of AMPK and Ca(2+)". American Journal of Physiology. Endocrinology and Metabolism. 282 (5): E1008-13. doi:10.1152/ajpendo.00512.2001. PMID 11934664. S2CID 7891060.
- ^ a b c Stoppani J, Hildebrandt AL, Sakamoto K, Cameron-Smith D, Goodyear LJ, Neufer PD (December 2002). "AMP-activated protein kinase activates transcription of the UCP3 and HKII genes in rat skeletal muscle". American Journal of Physiology. Endocrinology and Metabolism. 283 (6): E1239-48. doi:10.1152/ajpendo.00278.2002. PMID 12388122. S2CID 27188844.
- ^ a b Ojuka EO (May 2004). "Role of calcium and AMP kinase in the regulation of mitochondrial biogenesis and GLUT4 levels in muscle". The Proceedings of the Nutrition Society. 63 (2): 275–8. doi:10.1079/PNS2004339. hdl:11427/35025. PMID 15294043.
- ^ a b Winder WW, Holmes BF, Rubink DS, Jensen EB, Chen M, Holloszy JO (June 2000). "Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle". Journal of Applied Physiology. 88 (6): 2219–26. doi:10.1152/jappl.2000.88.6.2219. PMID 10846039. S2CID 5627995.
- ^ a b Ouchi N, Shibata R, Walsh K (April 2005). "AMP-activated protein kinase signaling stimulates VEGF expression and angiogenesis in skeletal muscle". Circulation Research. 96 (8): 838–46. doi:10.1161/01.RES.0000163633.10240.3b. PMID 15790954.
- ^ a b Hayashi T, Hirshman MF, Kurth EJ, Winder WW, Goodyear LJ (August 1998). "Evidence for 5' AMP-activated protein kinase mediation of the effect of muscle contraction on glucose transport". Diabetes. 47 (8): 1369–73. doi:10.2337/diabetes.47.8.1369. PMID 9703344.
- ^ a b Hayashi T, Hirshman MF, Fujii N, Habinowski SA, Witters LA, Goodyear LJ (April 2000). "Metabolic stress and altered glucose transport: activation of AMP-activated protein kinase as a unifying coupling mechanism". Diabetes. 49 (4): 527–31. doi:10.2337/diabetes.49.4.527. PMID 10871188.
- ^ a b Kurth-Kraczek EJ, Hirshman MF, Goodyear LJ, Winder WW (August 1999). "5' AMP-activated protein kinase activation causes GLUT4 translocation in skeletal muscle". Diabetes. 48 (8): 1667–71. doi:10.2337/diabetes.48.8.1667. PMID 10426389. S2CID 29891424.
- ^ a b c Merrill GF, Kurth EJ, Hardie DG, Winder WW (December 1997). "AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle". The American Journal of Physiology. 273 (6): E1107-12. doi:10.1152/ajpendo.1997.273.6.E1107. PMID 9435525.
- ^ a b c Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, et al. (October 2000). "Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia". Current Biology. 10 (20): 1247–55. doi:10.1016/S0960-9822(00)00742-9. PMID 11069105.
- ^ a b c Lee WJ, Kim M, Park HS, Kim HS, Jeon MJ, Oh KS, et al. (February 2006). "AMPK activation increases fatty acid oxidation in skeletal muscle by activating PPARalpha and PGC-1". Biochemical and Biophysical Research Communications. 340 (1): 291–5. doi:10.1016/j.bbrc.2005.12.011. PMID 16364253.
- ^ a b c Suwa M, Egashira T, Nakano H, Sasaki H, Kumagai S (December 2006). "Metformin increases the PGC-1alpha protein and oxidative enzyme activities possibly via AMPK phosphorylation in skeletal muscle in vivo". Journal of Applied Physiology. 101 (6): 1685–92. doi:10.1152/japplphysiol.00255.2006. PMID 16902066. S2CID 17877895.
- ^ a b Ojuka EO, Nolte LA, Holloszy JO (March 2000). "Increased expression of GLUT-4 and hexokinase in rat epitrochlearis muscles exposed to AICAR in vitro". Journal of Applied Physiology. 88 (3): 1072–5. doi:10.1152/jappl.2000.88.3.1072. PMID 10710405. S2CID 22504819.
- ^ Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E, et al. (August 2008). "AMPK and PPARdelta agonists are exercise mimetics". Cell. 134 (3): 405–15. doi:10.1016/j.cell.2008.06.051. PMC 2706130. PMID 18674809.
- ^ a b c Wang HJ, Lee CS, Yee RS, Groom L, Friedman I, Babcock L, et al. (October 2020). "Adaptive thermogenesis enhances the life-threatening response to heat in mice with an Ryr1 mutation". Nature Communications. 11 (1): 5099. Bibcode:2020NatCo..11.5099W. doi:10.1038/s41467-020-18865-z. PMC 7547078. PMID 33037202.
- ^ Stein SC, Woods A, Jones NA, Davison MD, Carling D (February 2000). "The regulation of AMP-activated protein kinase by phosphorylation". The Biochemical Journal. 345 (3): 437–43. doi:10.1042/0264-6021:3450437. PMC 1220775. PMID 10642499.
- ^ Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Mäkelä TP, et al. (2003). "Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade". Journal of Biology. 2 (4): 28. doi:10.1186/1475-4924-2-28. PMC 333410. PMID 14511394.
- ^ Hurst D, Taylor EB, Cline TD, Greenwood LJ, Compton CL, Lamb JD, Winder WW (October 2005). "AMP-activated protein kinase kinase activity and phosphorylation of AMP-activated protein kinase in contracting muscle of sedentary and endurance-trained rats". American Journal of Physiology. Endocrinology and Metabolism. 289 (4): E710-5. doi:10.1152/ajpendo.00155.2005. PMID 15928023. S2CID 25296834.
- ^ Hutber CA, Hardie DG, Winder WW (February 1997). "Electrical stimulation inactivates muscle acetyl-CoA carboxylase and increases AMP-activated protein kinase". The American Journal of Physiology. 272 (2 Pt 1): E262-6. doi:10.1152/ajpendo.1997.272.2.E262. PMID 9124333.
- ^ a b Taylor EB, Hurst D, Greenwood LJ, Lamb JD, Cline TD, Sudweeks SN, Winder WW (December 2004). "Endurance training increases LKB1 and MO25 protein but not AMP-activated protein kinase kinase activity in skeletal muscle". American Journal of Physiology. Endocrinology and Metabolism. 287 (6): E1082-9. doi:10.1152/ajpendo.00179.2004. PMID 15292028.
- ^ a b Taylor EB, Lamb JD, Hurst RW, Chesser DG, Ellingson WJ, Greenwood LJ, et al. (December 2005). "Endurance training increases skeletal muscle LKB1 and PGC-1alpha protein abundance: effects of time and intensity". American Journal of Physiology. Endocrinology and Metabolism. 289 (6): E960-8. doi:10.1152/ajpendo.00237.2005. PMID 16014350. S2CID 14454264.
- ^ Jørgensen SB, Treebak JT, Viollet B, Schjerling P, Vaulont S, Wojtaszewski JF, Richter EA (January 2007). "Role of AMPKalpha2 in basal, training-, and AICAR-induced GLUT4, hexokinase II, and mitochondrial protein expression in mouse muscle". American Journal of Physiology. Endocrinology and Metabolism. 292 (1): E331-9. doi:10.1152/ajpendo.00243.2006. PMID 16954334.
- ^ Röckl KS, Hirshman MF, Brandauer J, Fujii N, Witters LA, Goodyear LJ (August 2007). "Skeletal muscle adaptation to exercise training: AMP-activated protein kinase mediates muscle fiber type shift". Diabetes. 56 (8): 2062–9. doi:10.2337/db07-0255. PMID 17513699.
- ^ Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M (October 2007). "Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress". Cell Metabolism. 6 (4): 280–93. doi:10.1016/j.cmet.2007.08.011. PMID 17908557.
- ^ Winder WW (September 2001). "Energy-sensing and signaling by AMP-activated protein kinase in skeletal muscle". Journal of Applied Physiology. 91 (3): 1017–28. doi:10.1152/jappl.2001.91.3.1017. PMID 11509493. S2CID 18290292.
- ^ Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Müller C, Carling D, Kahn BB (January 2002). "Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase". Nature. 415 (6869): 339–43. Bibcode:2002Natur.415..339M. doi:10.1038/415339a. PMID 11797013. S2CID 4409274.
- ^ a b Sakamoto K, Göransson O, Hardie DG, Alessi DR (August 2004). "Activity of LKB1 and AMPK-related kinases in skeletal muscle: effects of contraction, phenformin, and AICAR". American Journal of Physiology. Endocrinology and Metabolism. 287 (2): E310-7. doi:10.1152/ajpendo.00074.2004. PMID 15068958.
- ^ Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, et al. (April 2004). "AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus". Nature. 428 (6982): 569–74. Bibcode:2004Natur.428..569M. doi:10.1038/nature02440. PMID 15058305. S2CID 4302317.
- ^ Taylor EB, Ellingson WJ, Lamb JD, Chesser DG, Winder WW (June 2005). "Long-chain acyl-CoA esters inhibit phosphorylation of AMP-activated protein kinase at threonine-172 by LKB1/STRAD/MO25". American Journal of Physiology. Endocrinology and Metabolism. 288 (6): E1055-61. doi:10.1152/ajpendo.00516.2004. PMID 15644453.
- ^ Hardie DG, Hawley SA (December 2001). "AMP-activated protein kinase: the energy charge hypothesis revisited". BioEssays. 23 (12): 1112–9. doi:10.1002/bies.10009. PMID 11746230. S2CID 39876683.
- ^ Thomson DM, Porter BB, Tall JH, Kim HJ, Barrow JR, Winder WW (January 2007). "Skeletal muscle and heart LKB1 deficiency causes decreased voluntary running and reduced muscle mitochondrial marker enzyme expression in mice". American Journal of Physiology. Endocrinology and Metabolism. 292 (1): E196-202. doi:10.1152/ajpendo.00366.2006. PMID 16926377.
- ^ a b Park, S. H.; Paulsen, S. R.; Gammon, S. R.; Mustard, K. J.; Hardie, D. G.; Winder, W. W. (December 2002). "Effects of thyroid state on AMP-activated protein kinase and acetyl-CoA carboxylase expression in muscle". Journal of Applied Physiology. 93 (6): 2081–2088. doi:10.1152/japplphysiol.00504.2002. ISSN 8750-7587. PMID 12433937.
- ^ Branvold, D. J.; Allred, D. R.; Beckstead, D. J.; Kim, H. J.; Fillmore, N.; Condon, B. M.; Brown, J. D.; Sudweeks, S. N.; Thomson, D. M.; Winder, W. W. (October 2008). "Thyroid hormone effects on LKB1, MO25, phospho-AMPK, phospho-CREB, and PGC-1alpha in rat muscle". Journal of Applied Physiology. 105 (4): 1218–1227. doi:10.1152/japplphysiol.00997.2007. ISSN 8750-7587. PMID 18669938. S2CID 2019764.
- ^ Winder, W. W.; Hardie, D. G.; Mustard, K. J.; Greenwood, L. J.; Paxton, B. E.; Park, S. H.; Rubink, D. S.; Taylor, E. B. (February 2003). "Long-term regulation of AMP-activated protein kinase and acetyl-CoA carboxylase in skeletal muscle". Biochemical Society Transactions. 31 (Pt 1): 182–185. doi:10.1042/bst0310182. ISSN 0300-5127. PMID 12546681.
- ^ Sun G, Tarasov AI, McGinty J, McDonald A, da Silva Xavier G, Gorman T, et al. (May 2010). "Ablation of AMP-activated protein kinase alpha1 and alpha2 from mouse pancreatic beta cells and RIP2.Cre neurons suppresses insulin release in vivo". Diabetologia. 53 (5): 924–36. doi:10.1007/s00125-010-1692-1. PMC 4306708. PMID 20221584.
- ^ Beall C, Piipari K, Al-Qassab H, Smith MA, Parker N, Carling D, et al. (July 2010). "Loss of AMP-activated protein kinase alpha2 subunit in mouse beta-cells impairs glucose-stimulated insulin secretion and inhibits their sensitivity to hypoglycaemia". The Biochemical Journal. 429 (2): 323–33. doi:10.1042/BJ20100231. PMC 2895783. PMID 20465544.
- ^ Claret M, Smith MA, Batterham RL, Selman C, Choudhury AI, Fryer LG, et al. (August 2007). "AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons". The Journal of Clinical Investigation. 117 (8): 2325–36. doi:10.1172/JCI31516. PMC 1934578. PMID 17671657.
- ^ Beall C, Hamilton DL, Gallagher J, Logie L, Wright K, Soutar MP, et al. (September 2012). "Mouse hypothalamic GT1-7 cells demonstrate AMPK-dependent intrinsic glucose-sensing behaviour". Diabetologia. 55 (9): 2432–44. doi:10.1007/s00125-012-2617-y. PMC 3411292. PMID 22760787.
- ^ Alquier T, Kawashima J, Tsuji Y, Kahn BB (March 2007). "Role of hypothalamic adenosine 5'-monophosphate-activated protein kinase in the impaired counterregulatory response induced by repetitive neuroglucopenia". Endocrinology. 148 (3): 1367–75. doi:10.1210/en.2006-1039. PMID 17185376.
- ^ McCrimmon RJ, Shaw M, Fan X, Cheng H, Ding Y, Vella MC, et al. (February 2008). "Key role for AMP-activated protein kinase in the ventromedial hypothalamus in regulating counterregulatory hormone responses to acute hypoglycemia". Diabetes. 57 (2): 444–50. doi:10.2337/db07-0837. PMID 17977955.
- ^ Fan X, Ding Y, Brown S, Zhou L, Shaw M, Vella MC, et al. (June 2009). "Hypothalamic AMP-activated protein kinase activation with AICAR amplifies counterregulatory responses to hypoglycemia in a rodent model of type 1 diabetes". American Journal of Physiology. Regulatory, Integrative and Comparative Physiology. 296 (6): R1702-8. doi:10.1152/ajpregu.90600.2008. PMC 2692788. PMID 19357294.
- ^ Li M, Zhang CS, Zong Y, Feng JW, Ma T, Hu M, et al. (September 2019). "Transient Receptor Potential V Channels Are Essential for Glucose Sensing by Aldolase and AMPK". Cell Metabolism. 30 (3): 508–524.e12. doi:10.1016/j.cmet.2019.05.018. PMC 6720459. PMID 31204282.
- ^ a b c Jia J, Bissa B, Brecht L, Allers L, Choi SW, Gu Y, et al. (March 2020). "AMPK, a Regulator of Metabolism and Autophagy, Is Activated by Lysosomal Damage via a Novel Galectin-Directed Ubiquitin Signal Transduction System". Molecular Cell. 77 (5): 951–969.e9. doi:10.1016/j.molcel.2019.12.028. PMC 7785494. PMID 31995728.
- ^ Papadopoulos C, Kirchner P, Bug M, Grum D, Koerver L, Schulze N, et al. (January 2017). "VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy". The EMBO Journal. 36 (2): 135–150. doi:10.15252/embj.201695148. PMC 5242375. PMID 27753622.
- ^ a b c Jia J, Claude-Taupin A, Gu Y, Choi SW, Peters R, Bissa B, et al. (January 2020). "Galectin-3 Coordinates a Cellular System for Lysosomal Repair and Removal". Developmental Cell. 52 (1): 69–87.e8. doi:10.1016/j.devcel.2019.10.025. PMC 6997950. PMID 31813797.
- ^ Grebe A, Latz E (March 2013). "Cholesterol crystals and inflammation". Current Rheumatology Reports. 15 (3): 313. doi:10.1007/s11926-012-0313-z. PMC 3623938. PMID 23412688.
- ^ Chauhan S, Kumar S, Jain A, Ponpuak M, Mudd MH, Kimura T, et al. (October 2016). "TRIMs and Galectins Globally Cooperate and TRIM16 and Galectin-3 Co-direct Autophagy in Endomembrane Damage Homeostasis". Developmental Cell. 39 (1): 13–27. doi:10.1016/j.devcel.2016.08.003. PMC 5104201. PMID 27693506.
- ^ Yue Y, Nabar NR, Shi CS, Kamenyeva O, Xiao X, Hwang IY, et al. (September 2018). "SARS-Coronavirus Open Reading Frame-3a drives multimodal necrotic cell death". Cell Death & Disease. 9 (9): 904. doi:10.1038/s41419-018-0917-y. PMC 6125346. PMID 30185776.
- ^ Zhang CS, Li M, Ma T, Zong Y, Cui J, Feng JW, et al. (October 2016). "Metformin Activates AMPK through the Lysosomal Pathway". Cell Metabolism. 24 (4): 521–522. doi:10.1016/j.cmet.2016.09.003. PMID 27732831.
- ^ a b Merrill GM, Kurth E, Hardie DG, Winder WW (2020). "AMP-Activated Protein Kinase: Friend or Foe in Cancer?". Annual Review of Cancer Biology. 4: 1–16. doi:10.1146/annurev-cancerbio-030419-033619.
- ^ Schneider, Carolin; Hilbert, Jorina; Genevaux, Franziska; Höfer, Stefanie; Krauß, Lukas; Schicktanz, Felix; Contreras, Constanza Tapia; Jansari, Shaishavi; Papargyriou, Aristeidis; Richter, Thorsten; Alfayomy, Abdallah M.; Falcomatà, Chiara; Schneeweis, Christian; Orben, Felix; Öllinger, Ruppert (2024-06-17). "A Novel AMPK Inhibitor Sensitizes Pancreatic Cancer Cells to Ferroptosis Induction". Advanced Science. doi:10.1002/advs.202307695. ISSN 2198-3844. PMC 11336956.
External links
[edit]- AMP-activated+protein+kinase at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- EC 2.7.11.31